|
|
Optimization of coarse grained molecular simulation
Primary tabs
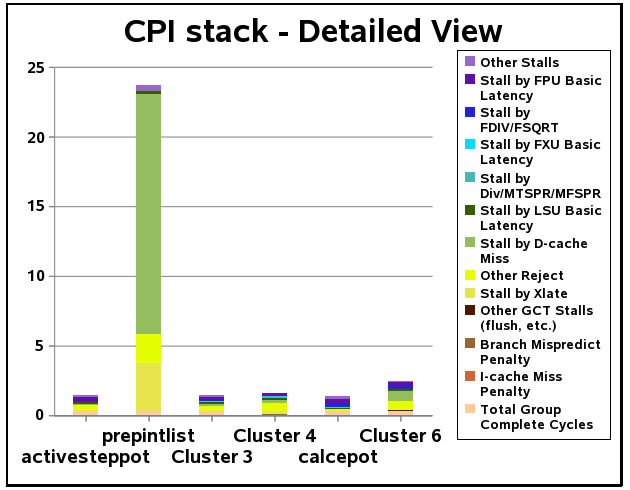
Molecular simulations based on coarse grained methods, can significantly reduce the time to compute flexible models of proteins, but with enough quality to improve docking experiments. We are currently optimizing two software applications (dpEDMD and DISCRETE) to scale the problems on a larger number of cores. With faster simulations we will be able to scan larger conformational space for docking (protein-ligand and protein-protein) experiments.