|
|
Platform's capacities
Primary tabs
Platform's capacities

We can process patients’ data to map genetic variability of different phenotypes (e.g. disease patient vs control patients). Our software workflow can read SNP-microarray data from different platforms (Affymetrix, Illumina, etc.) that can normally map only a part of human variability (around 500,000 selected SNPs) and expand it to the Whole Genome. Our application will use the 1000G database to fill the missing parts of the genome through imputation. This is a complex and heavy-computation step that has two advantages: i) it expands the identification of genetic factors to the whole genome and ii) it enables multi-platform comparison.
We have developed a novel methodology to perform genome-wide association studies (GWAS), aimed at detecting associations between common genetic variants and human complex traits and diseases. This methodology is composed of several stages, including i) an initial filtering of genetic data from the control and individual cases, ii) haplotype phase inference from genotype data, iii) genotype imputation using several reference panels, iv) analysis of single SNP association, and many others.
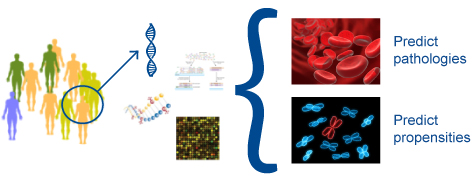
Identification of genetic factors associated to diseases is an important step toward understanding the biological mechanisms of the pathologies and identifying future drug targets. This information is very important for patient early diagnosis (predicting pathology) and prognosis. We can process genomic data of patients to train and build predictive models based on machine learning techniques, and support the development of future diagnostic and prognostic kits.

Genetic variants in drug targets can induce drug resistance. The cost of testing a single mutant using in vitro or in vivo experiments is unaffordable for highly variable targets (e.g. virus proteins). When experimental and clinical data is available, machine learning methods can be used to model drug resistance response, but the reliability of predictions is very poor for untrained scenarios. Our approach is to perform high-accuracy prediction of affinities between disease-targets and drugs based on molecular simulation. There are a number of techniques that we can combine for these calculations: Ensemble docking, PELE with docking, and molecular mechanics simulation with Poisson-Boltzmann.
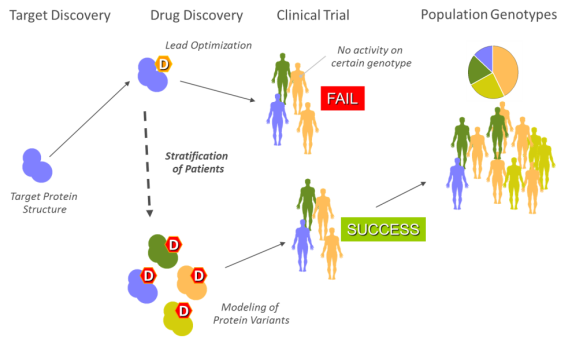
We can model and scan new targets (protein genotypes) with ligand-based and structure-based drug discovery strategies. Ligand-based strategy assumes the hypothesis that similar molecules are likely to share functionality. Our platform allows a target-fit library enrichment using 2D-QSAR, Docking and hybrid Docking/QSAR strategies that can be later be refined and experimentally validated to identify new lead candidates.