|
|
Medicahead's technologies
Primary tabs
Software Applications
Website: PELE
Using technological advances in protein structure prediction, we have developed PELE (Protein Energy Landscape Exploration), a novel method for performing protein energy landscape explorations. PELE combines a Monte Carlo stochastic approach with protein structure prediction algorithms and is capable of accurately reproducing long time scale processes in only few hours of CPU. The Figure below shows the heuristic algorithm for the landscape exploration method which is based on three main steps: an initial perturbation, a side chain sampling and a final minimization.
Website: PyDock
Rigid-body docking and scoring by pyDock. pyDockWEB is a web server for the structural prediction of protein-protein interactions. Given the 3D coordinates of two interacting proteins, pyDockWEB returns the best rigid-body docking orientations generated by FTDock and evaluated by pyDock scoring function, which includes electrostatics, desolvation energy and limited van der Waals contribution.
Website: PMUT
Pmut is a software aimed at the annotation and prediction of pathological mutations, and in particular at answering the following question: given a mutation happening at a specific location in a protein sequence, can we say whether it will be pathological (that is, a mutation that can lead to disease for the carrier) or non-pathological/neutral (no effect on the carrier's health)? Pmut is based on the use of different kinds of sequence information to label mutations, and neural networks to process this information (Ferrer-Costa et al., 2004). It provides a very simple output: a yes/no answer and a reliability index.

HT-GWASi is a High Througput pipeline to analyze genetic factor of complex diseases.

SEABED is an application to scan new large libraries of compounds using chemoinformatics and docking strategies. Ligand-based strategy assumes the hypothesis that similar molecules are likely to share functionality. Therefore small molecules sharing structural descriptors with known drugs are likely to share the same activity against the disease-target. Using this principle we will implement a quantitative structure-activity relationship (2D-QSAR) module to enrich libraries for a virtual screening of hits (using a rigid docking approach). The Docking/QSAR hybrid strategy will be a powerful tool to move from a large list of molecules (millions) to a short sub-set of lead compounds.
Software Stack
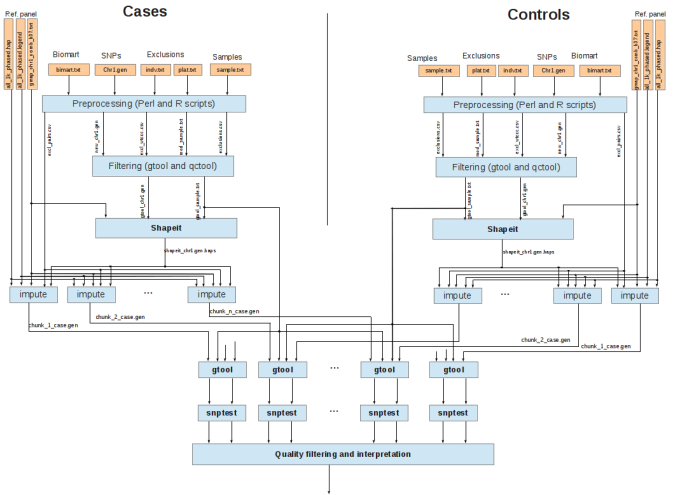
The Genome-Wide Analysis Study workflow runs in a workflow fashion, with multiple modules having different requirements of performance, parallelism, memory usage, etc. Due to the computationally intensive work involved in each stage and the huge amount of information to be processed, it is necessary to develop an efficient implementation of our GWAS methodology. To reach this goal, we will develop a preliminary implementation of the GWAS workflow using the COMP Superscalar framework. COMPSs will be useful to exploit the inherent parallelism of applications at execution time. The results of these studies will be very useful to take decisions related to the optimization of some stages, improvement of some algorithms and possible implementations using state-of-the-art architectures like GPUs.
Optimization of coarse grained molecular simulation
Molecular simulations based on coarse grained methods can significantly reduce the time necessary to compute flexible models of proteins, while doing so with enough quality to improve docking experiments. We are currently optimizing two software applications (dpEDMD and DISCRETE) to scale the problems on a larger number of cores. With faster simulations we will be able to scan larger conformational space for docking (protein-ligand and protein-protein) experiments.
Structure Based Drug Discovery (SBDD) is based on the rational design of small molecules that can lock (activate or inhibit) the active site of the target protein. Realistic models of the protein are necessary to increase the success rate of drug candidates. Molecular Dynamics and coarse-grained molecular simulation methods are a powerful method to build flexible models, but the computational cost and the size of the data is difficult to manage for large projects. We are currently developing new high-scalable data frameworks for simulations, based on non-relational and non-centralized data store systems.
Molecular simulations based on coarse grained methods, can significantly reduce the time to compute flexible models of proteins, but with enough quality to improve docking experiments. We are currently optimizing two software applications (dpEDMD and DISCRETE) to scale the problems on a larger number of cores. With faster simulations we will be able to scan larger conformational space for docking (protein-ligand and protein-protein) experiments.

We are working to optimize genomics applications in the supercomputing environment for large genomics projects. Among our relevant projects is the ongoing preparation of a genomic workflow analysis to be deployed in a cloud environment, being set in collaboration with BSC’s Grid computing and Clusters team. We are also establishing a prototype for Data Distribution Center for the genomics projects.
Patents
PCT Application Nº PCT/EP2012/069636
Applicant:
- Universitat de Barcelona
- Barcelona Supercomputing Center - Centro Nacional de Supercomputación
- Fundació Privada Institut de Recerca Biomèdica
- The University of Nottingham
Title:
- Method of Exploring the Flexibility of Macromolecular Targets and its use in Rational Drug Design
|
|
|
 |