
Alfonso Valencia ha liderado este proyecto para descubrir la ubicación de los cambios y su relación com la diversidad celular i el origen del cáncer.

Un equipo de científicos de distintas instituciones liderados por el investigador del BSC-CNS Alfonso Valencia y por el investigador de la Universidad de Newcastle Daniel Rico ha desarrollado un método para detectar las zonas del epigenoma donde se originan los cambios que dan lugar a la diversidad celular y que también pueden estar vinculadas con el origen y desarrollo del cáncer. En el estudio, que ha sido publicado en Nucleic Acids Research, se ha desarrollado un método computacional que ha sido aplicado en la hematopoyesis, el proceso de formación de la sangre a partir de células madre.
El sistema permite integrar diferentes datos epigenómicos para clasificar diferentes tipos de muestras y extraer de manera automática aquellas regiones del genoma donde se van a producir los cambios químicos que afectan a la diversidad celular. Asimismo, relacionan estas regiones y sus cambios químicos con el origen y desarrollo de diferentes leucemias.
El genoma humano es una secuencia de letras inerte que necesita de la colaboración del epigenoma para cobrar vida. El genoma es como un libro donde todas las letras van seguidas, sin espacios, puntos o comas. Este libro sería difícil de leer e imposible de comprender. Sin embargo, al colocar correctamente los signos de puntuación (comas, puntos, signos de interrogación, etc) podemos leer y entender perfectamente esa sucesión inerte de letras. Esto es lo que hace el epigenoma, son cambios químicos que nos permiten comprender cómo hay que leer e interpretar correctamente el genoma.
Por ello, el estudio del epigenoma se ha convertido en una pieza clave para entender cómo de una única célula común se desarrollan la gran diversidad de células que componen nuestros órganos y tejidos, o cómo una célula sana puede acabar transformándose en tumoral. Sin embargo, y a pesar de las grandes colecciones de datos disponibles hasta el momento, localizar aquellas regiones del genoma con cambios químicos (signos de puntuación) y el tipo de cambio sigue siendo un reto para la comunidad científica.
Estos cambios químicos se producen a lo largo del desarrollo y como respuesta a factores externos pudiendo llevar a la aparición de enfermedades. Por ello, identificar estos biomarcadores epigenéticos es imprescindible para crear nuevas herramientas que ayuden en el diagnóstico y tratamiento de distintas enfermedades.
Los estudios moleculares clasifican a los pacientes usando la expresión de los genes como biomarcadores mirando si están encendidos o apagados. El método propuesto en este trabajo permite identificar aquellas regiones que van a regular como interruptores biológicos este encendido y apagado de los genes, que podrían ser usados como biomarcadores epigenómicos que complementen las clasificaciones moleculares actuales.
“El desarrollo de este tipo de metodologías es muy importante porque hasta ahora se habían estudiado las diferencias de los distintos tipos celulares y tejidos a nivel de qué genes están encendidos o apagados, el producto final, pero no se conocía demasiado sobre cuáles son los interruptores para apagarlos o encenderlos (epigenoma). Esta información es imprescindible para poder abordar con éxito nuevas terapias que permitan apretar los interruptores adecuados cuando la célula pierde el control desarrollando enfermedades como el cáncer. Entender esta regulación nos llevará un paso más allá en la aplicación de la medicina personalizada ”, comenta Enrique Carrillo, primer coautor del trabajo e investigador del CNIO.
Por otro lado, el esfuerzo desarrollado por los proyectos englobados en IHEC (Consorcio Internacional del Epigenoma Humano) durante los últimos años ha permitido generar una enorme colección de datos que nos permite profundizar en el conocimiento de la regulación celular y sus alteraciones. “Los cerca de 9000 epigenomas humanos disponibles en IHEC junto con las últimas actualizaciones realizadas en el supercomputador MareNostrum 4 van a permitirnos desarrollar nuevos sistemas de análisis y métodos computacionales más complejos que permitan profundizar y acelerar la adquisición de nuevos conocimientos para el manejo de la salud humana, no sólo en términos de diagnóstico y tratamiento de distintas enfermedades, sino también en estilos de vida saludables” comenta Alfonso Valencia, Director del Departamento de Ciencias de la Vida del BSC-CNS.
Los métodos y resultados obtenidos en este trabajo son de acceso público sin restricciones y permitirán a la comunidad científica la identificación de nuevos biomarcadores epigenéticos que podrán ser utilizados como parte de las herramientas de diagnóstico y tratamiento en Medicina Personalizada de Precisión.
En este proyecto han intervenido investigadores del Centro Nacional de Investigaciones Oncológicas (CNIO), del Instituto de Biología Evolutiva (IBE:CSIC-UPF), del Barcelona Supercomputing Center – Centro Nacional de Supercomputación (BSC-CNS), del Institut d'Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS) y del Institute of Cellular Medicine (Newcastle University). Este estudio ha sido financiado por la Unión Europea (FP7/2007–2013, 282510; BLUEPRINT), el Ministerio de Economía y Competitividad (BFU2015–71241-R) y la iniciativa filantrópica “Amigos del CNIO”.
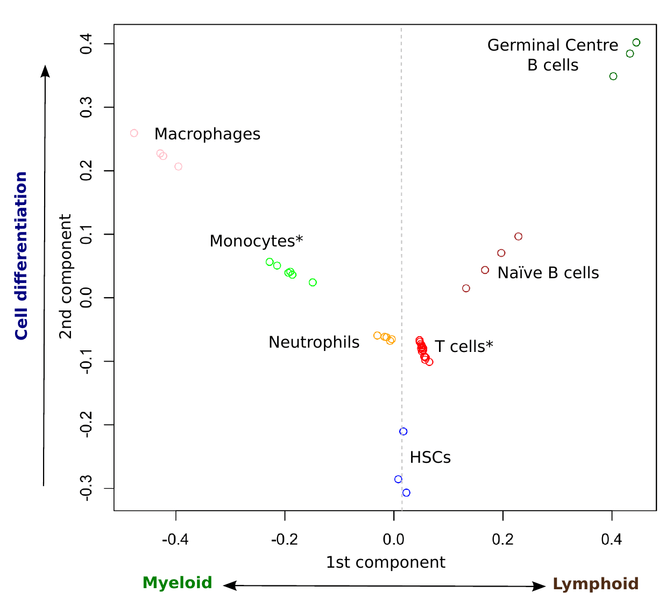
Figura: Distribución de muestras sanguíneas según sus perfiles epigenómicos producida con el nuevo método computacional.
Artículo de referencia:
Enrique Carrillo-de-Santa-Pau, David Juan, Vera Pancaldi, Felipe Were, Ignacio Martin-Subero, Daniel Rico, Alfonso Valencia, on behalf of The BLUEPRINT Consortium; Automatic identification of informative regions with epigenomic changes associated to hematopoiesis. Nucleic Acids Res 2017 gkx618. doi: 10.1093/nar/gkx618