|
|
Scaling of GWAS with imputation using COMP Superscalar
Primary tabs
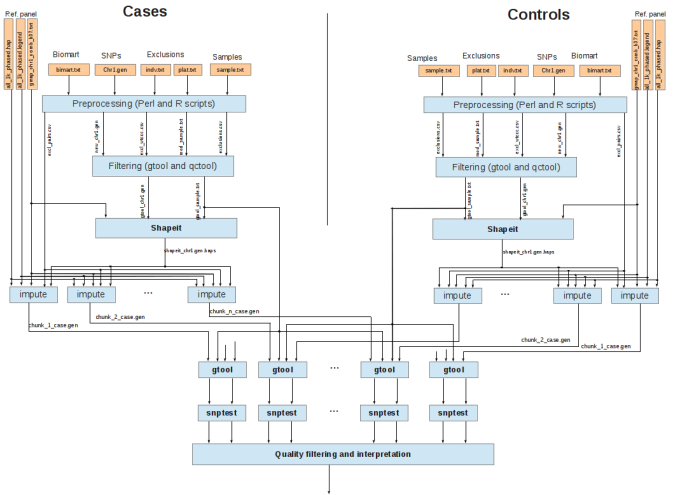
The Genome-Wide Analysis Study workflow runs in a workflow fashion, with multiple modules having different requirements of performance, parallelism, memory usage, etc. Due to the computationally intensive work involved in each stage and the huge amount of information to be processed, it is necessary to develop an efficient implementation of our GWAS methodology. To reach this goal, we will develop a preliminary implementation of the GWAS workflow using the COMP Superscalar framework. COMPSs will be useful to exploit the inherent parallelism of applications at execution time. The results of these studies will be very useful to take decisions related to the optimization of some stages, improvement of some algorithms and possible implementations using state-of-the-art architectures like GPUs.
Optimization of coarse grained molecular simulation
Molecular simulations based on coarse grained methods can significantly reduce the time necessary to compute flexible models of proteins, while doing so with enough quality to improve docking experiments. We are currently optimizing two software applications (dpEDMD and DISCRETE) to scale the problems on a larger number of cores. With faster simulations we will be able to scan larger conformational space for docking (protein-ligand and protein-protein) experiments.